MED | LHS杜洋團隊在Nature Communications揭示人體孤兒受體GPR88激活和別構調節機制
G蛋白偶聯受體(G-protein coupled receptor,GPCR)是人類基因組中最大的膜信號蛋白家族,包含800多個成員[1]。然而其中大約140個受體的內源性配體仍然未被確定[2,3]。這些孤兒受體(orphan GPCR,oGPCR)的配體結合和信號傳導機理尚不明確,例如自激活機制。因此,為oGPCR“脫孤”以及解析它們的結構能夠幫助研究者進一步了解GPCR的分子機理,為相關疾病的藥物開發帶來新的思路。
GPR88隸屬于A類GPCR,在大腦(特別是紋狀體)中特異性表達[2,3,5]。該受體能夠調節GABA和谷氨酸信號通路,以及包括多巴胺受體和阿片類受體等一些其他的GPCR活性[6,7]。轉錄分析和小鼠敲除研究表明,GPR88在調節大腦和行為功能(例如認知,情緒,基于獎勵的學習和運動控制)中起著重要作用[6, 8, 9]。因此,GPR88是治療中樞神經系統相關疾病(包括精神分裂癥,帕金森氏病,躁郁癥,焦慮,抑郁癥和成癮)的潛在藥物靶標[2, 3, 10]。
近日,香港中文大學(深圳)醫學院、科比爾卡創新藥物開發研究院杜洋教授、柳正教授團隊聯合埃爾朗根-紐倫堡大學Peter Gmeiner教授團隊和日本東北大學Asuka Inoue教授團隊在國際一流科學期刊Nature Communication雜志發表了題為“Activation and allosteric regulation of the orphan GPR88-Gi1 signaling complex”的最新研究成果。該研究基于冷凍電鏡技術解析了人類GPR88-Gi1和合成激動劑(1R,2R)-2-PCCA信號復合物的結構,表明了(1R,2R)-2-PCCA結合在由跨膜螺旋5、6的細胞質末端和Gi1的α5螺旋C末端組成的變構位點,展示了正構結合位點的電子密度代表未知的內源性配體。MD和突變研究揭示了一組獨特的結構特征和水介導的極性網絡的GPR88的獨特激活機制,為理解GPR88的配體結合、激活和信號傳導機制提供了一個結構框架,并將促進神經精神疾病的創新藥物發現以及該受體的“脫孤”。香港中文大學(深圳)為第一作者單位,杜洋教授為該研究最后通訊作者。陳耕博士、徐俊博士、Maximilian Schmidt和Asuka Inoue并列第一作者。

點擊圖片,閱讀全文
在結合或不結合2-PCCA的情況下,研究者將GPR88-Gi1復合物與scFv16(用于穩定Gi的抗體)組裝在一起,獲得了兩個復合物在2.4 ?和3.0??分辨率下的cryo-EM密度圖,并藉此重建了原子結構模型(圖1)。受體的N端,C端,細胞內環3(ICL3),細胞外環1(ECL1)和細胞外環2(ECL2)的電子密度有所缺失,表明這些區域較為靈活和無序。與近期解析的GPR52不同,GPR88的ECL2沒有良好折疊并扮演自激活的激動劑[4],正構位點未確認的密度表明其可能擁有獨特的自激活機制。2-PCCA則結合在變構位點,其周邊推測存在三個膽固醇分子。
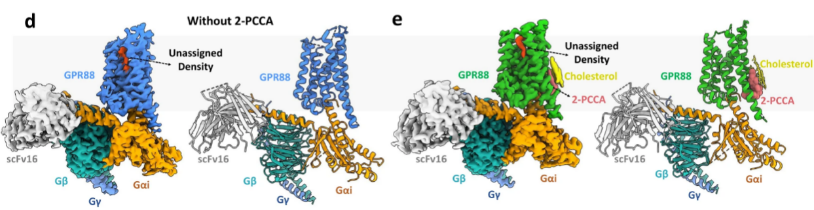
圖1 GPR88-Gi1冷凍電鏡結構
(圖源:Chen G, et al., Nat Commun, 2022)
GPR88的正構位點由TM3、TM4、TM5、TM7和ECL2的一部分組成,在A類GPCR中較為保守。對該口袋的表面靜電勢分析表明,TM3、TM4和TM5產生了一個長的疏水孔,同時細胞外表面帶電并且親水(圖2)。結合兩極化的口袋特征和位于口袋中電子密度的形狀,作者推測該電子密度代表了一種脂質分子,扮演內源性配體的角色。該脂質分子非極性尾部插入疏水孔中,極性頭部則位于口袋的細胞外側。
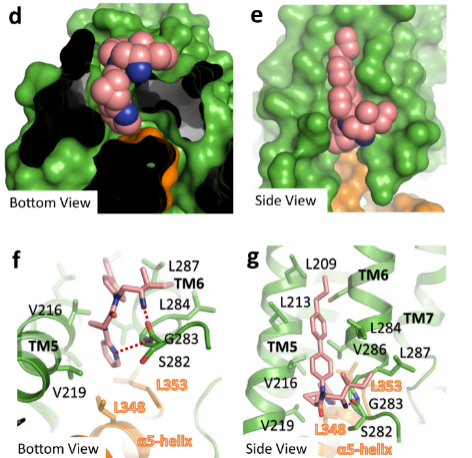
圖2 GPR88的變構結合口袋
(圖源:Chen G, et al., Nat Commun, 2022)
GPR88的變構位點由TM5、TM6的細胞質末端和Gi1的α5螺旋C末端組成,該變構位點未在其他GPCR結構中報導過。結合在該口袋的2-PCCA的中心酰胺有三個取代基,分別為氨烷基(aminoalkyl,R1),吡啶基環丙基(pyridylcyclopropyl,R2)和聯芳基(biaryl,R3)。其中R2插入了口袋,芳環中的鄰位氮與甘氨酸的主鏈 NH 形成氫鍵;R1、R3和TM5、TM6面向膜的表面疏水氨基酸(包括纈氨酸,異亮氨酸,亮氨酸和半胱氨酸)形成了廣泛的疏水相互作用。研究者還觀察到三個假定的膽固醇分子和TM5、TM6一起形成了和正構位點相似的疏水孔,進一步增強了2-PCCA在變構位點的結合。構效關系分析結果證明:配體R2基團的吡啶和變構口袋表面亮氨酸、纈氨酸(疏水氨基酸)對2-PCCA結合變構位點并激活受體非常重要。
GPR88的活性構象和視紫紅質(rhodopsin)等其他A類GPCR有所不同。首先,GPR88較短的跨膜螺旋、更靠內的TM6,使得TM5和TM6細胞質末端界面形成空腔,即變構位點。其次,序列對比表明GPR88的ECL上缺少半胱氨酸,進而缺少二硫鍵的形成,導致GPR88的ECL更加動態。另外,GPR88缺少A類GPCR保守的撥動開關W6.48和P5.50-I/L3.40-F6.44基序。撥動開關W6.48在其他視紫紅質家族的GPCR激活中觸發TM6向外移動,GPR88中該位點則被蘇氨酸代替。P-I/L-F基序的空間重排在從細胞外域到G蛋白耦合界面構象變化的傳播中起關鍵作用,然而在GPR88中,P5.50被Q2045.50所取代并和L1283.40、F6.44相互作用(即Q-L-F基序);同時,L1283.40位于未確認的電子密度下方并可能和假定的內源性配體有直接接觸。將Q2045.50突變為脯氨酸(P)增強了2-PCCA對GPR88的活性,證明在信號傳導上,更典型的P-L-F基序比GPR88的Q-L-F基序可能更加有效(圖3)。
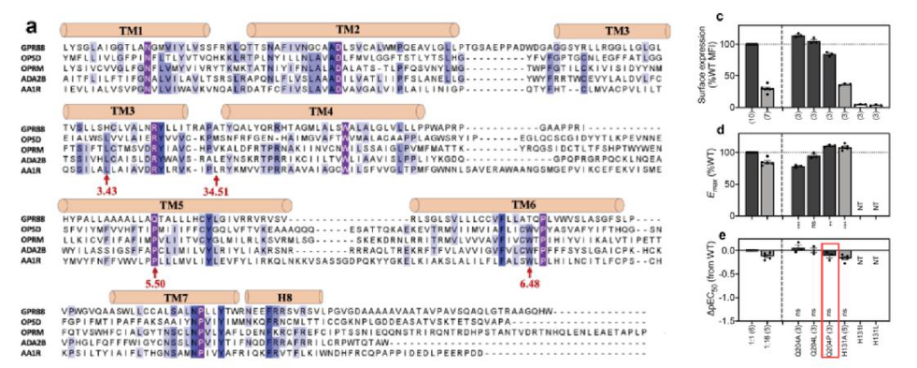
圖3 序列比對,Q2045.50和H1313.43的突變實驗
(圖源:Chen G, et al., Nat Commun, 2022)
TM7中的N7.49P7.50xxY7.53基序是另一個在A類GPCR中保守的序列。在活性視紫紅質和μ阿片受體(μOR)中,Y7.53和N7.49和TM3及TM5通過水形成極性網絡相互作用,其中的氫鍵涉及骨架羰基[11, 12]。在GPR88中,親水氨基酸組氨酸(H1313.43)直接和Y3367.53形成氫鍵。值得注意的是,研究者觀察到位于Y3367.53和H1313.43界面上兩個水分子的電子密度(圖4)。上方的水分子進一步增強了N7.49P7.50xxY7.53基序和TM3之間的極性相互作用;下方的水分子則介導了與視紫紅質和μ阿片受體中相似的,Y3367.53、H1313.43和Y2125.58之間的氫鍵網絡。
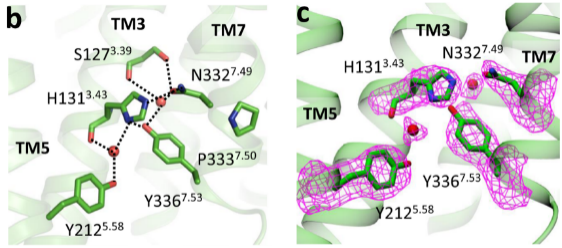
圖4 水介導的極性網絡
(圖源:Chen G, et al., Nat Commun, 2022)
研究者此后又進行了從活性狀態到非活性狀態(通過刪除2-PCCA、Gi1和scFv16)的元動力學模擬(metadynamics simulations),結果和觀察到的結構特征相符:撥動開關T6.48的2.2 ?位移;Q5.50-L3.40-F6.44基序的大型結構重排;水介導的氫鍵網絡進行了重新排列:H1313.43下方的水分子從受體核心移位,導致N7.49P7.50xxY7.53的構象變化;上方的水分子仍然存在,介導TM3和TM7之間的一個小極性網絡。值得注意的是,H1313.43在未被激活時也參與了該極性網絡,表明了其在穩定GPR88的非活性構象中的潛在作用。值得注意的是,受體失活重塑了變構位點。結果是在模擬中,TM6的向內運動縮小了變構位點的疏水袋,阻止了2-PCCA的結合(圖5)。
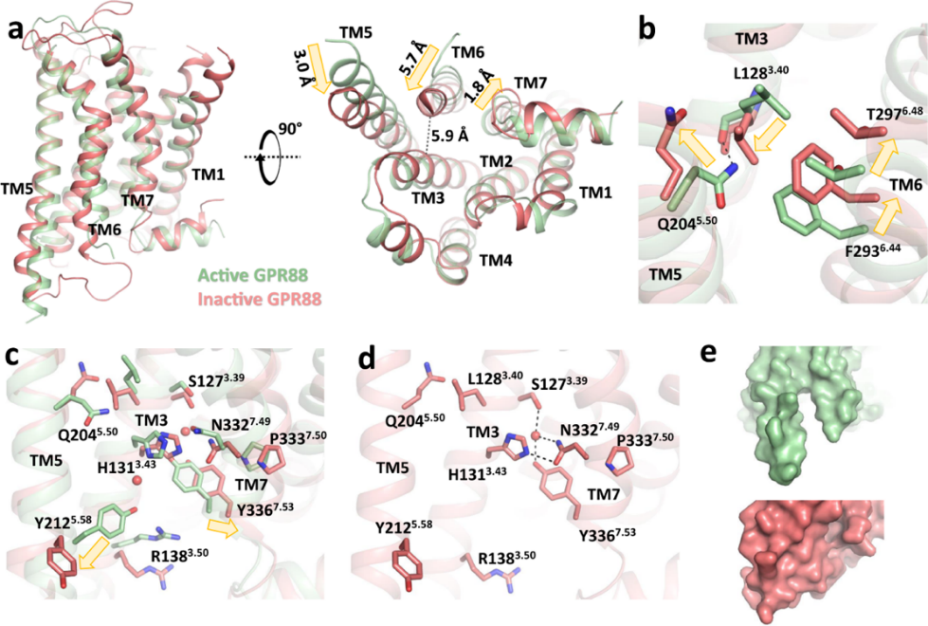
圖5非活性狀態GPR88的元動力學模擬
(圖源:Chen G, et al., Nat Commun, 2022)
結合或不結合2-PCCA的結構在G蛋白耦合界面幾乎相同,在2-PCCA結合結構中G蛋白的略微向上移動。與先前報道的復合物結構相似,α5螺旋的C末端插入由TM3和TM5-7的細胞質末端形成的空腔中。該空腔中,α5螺旋和TM3、TM5和TM6上的疏水氨基酸通過疏水接觸相互作用,同時α5螺旋半胱氨酸的骨架羰基和TM3的精氨酸之間形成了氫鍵。值得注意的是,變構2-PCCA還通過與GPR88的TM5-TM6、Gi1的α5螺旋參與疏水網絡,進一步穩定GPR88-Gi1信號傳導復合物的界面并且導致Gi1的輕微移動。
除了疏水接觸外,TM5的細胞質末端和α5螺旋的底部之間存在極性相互作用, GPR88的ICL2和Gi1的αN螺旋之間也存在一個極性界面,主要涉及氫鍵的形成。這些極性相互作用對于GPR88和Gi1之間的耦合可能至關重要(圖6)。
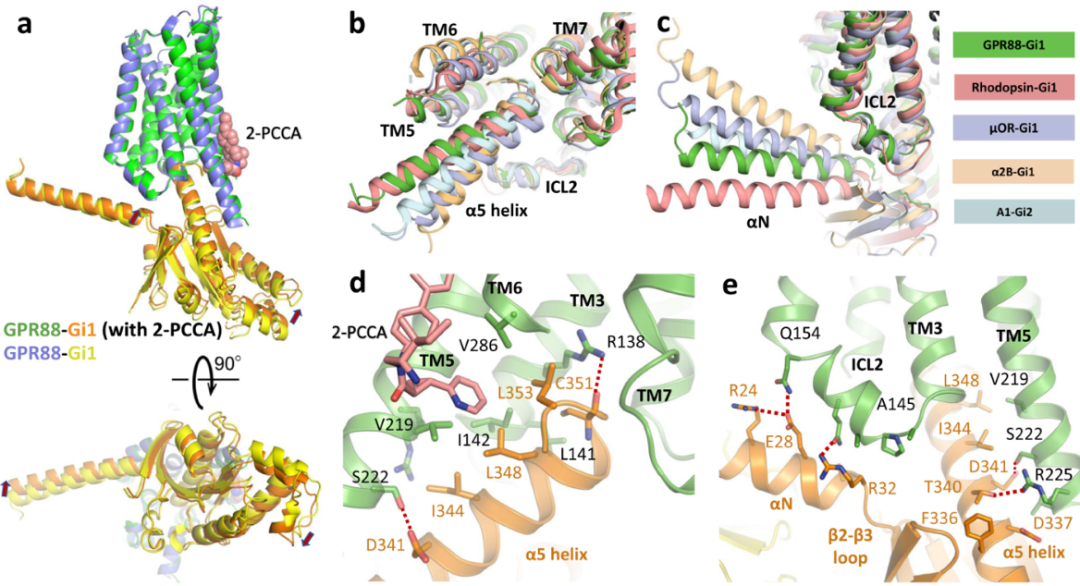
圖6 GPR88和Gi1的耦合界面
(圖源:Chen G, et al., Nat Commun, 2022)
綜上所述,該研究展示了結合或不結合合成激動劑(2-PCCA)的GPR88-Gi1信號傳導復合物結構。這些結構在GPR88的規范正構袋中揭示了相似的電子密度,該密度可能代表受體的假定內源配體。2-PCCA是一種變構激動劑,與變構位點結合,直接涉及與G蛋白的相互作用,進一步穩定了信號傳導復合物,從而促進了GPCR88的高活性。Apo-GPR88-Gi和2-PCCA-GPR88-Gi的高分辨率結構對比表明,正構袋中的假定內源配體可以與GPR88共純化。此外,未確定的密度和正位口袋的特性表明GPR88可能是響應某些生物活性脂質的受體。令人感興趣的是,與其內源性激動劑S1P結合的鞘氨醇1-磷酸(S1P)受體的最新結構顯示出與GPR88相似的正位結合口袋,口袋中形成了用于結合S1P的穿透性長隧道[13]。因此研究者推測,GPR88的內源性激動劑可能是一種與S1P相似的脂質分子,并且該脂質配體可能能夠與GPR88的變構位點結合以調節信號傳導。但是,作者不能排除脂質分子具有分支結構和未確定的電子密度由靈活區形成的可能性。
該研究還揭示了一個廣泛的由水介導的氫鍵網絡,該網絡連接了受體細胞外結構域和G蛋白,穩定GPR88的活性構象。GPR88跨膜核心區的不保守殘基H1313.43不僅在介導極網絡中扮演著關鍵作用,而且還是維持GPR88的功能表達的關鍵,這揭示此孤兒受體中獨特的結構特征。通過比較活性cryo-EM結構與元動力學生成的GPR88的去活化模型,該研究揭示了與GPR88激活相關的關鍵構象變化。
總而言之,該研究提供了理解孤兒受體GPR88的配體結合,激活和信號轉導的結構基礎。這些發現將促進GPR88的“脫孤”。基于結構的激動劑、拮抗劑設計可能會為中樞神經系統疾病提供有價值的候選藥物。
?
第一作者簡介

陳耕 博士
香港中文大學(深圳)醫學院/科比爾卡創新藥物開發研究院助理研究員,鵬城孔雀計劃特聘崗位。博士畢業于Baylor University,并在UT Austin從事博士后研究工作,現從事GPCR藥物靶點的結構研究和藥物開發。
?

杜洋 教授
醫學院助理院長(科研與創新)
香港中文大學(深圳)醫學院助理院長(科研與創新)、科比爾卡創新藥物開發研究院研究員,博士生導師,教育部青年,廣東省珠江青年拔尖人才。師從諾貝爾化學獎得主Brian Kobilka教授從事GPCR相關的博士后研究工作,回國前獲得密歇根大學安娜堡醫學院tenure-track助理教授職位等。迄今已發表約60篇高質量SCI論文,包括以第一或通訊作者(含共同)在Cell、Sci. Adv.、JACS、Nature Comm.、Cell Res.等國際一流期刊報道的科研和轉化成果。擬招聘博士后和學術型碩士/博士生,歡迎感興趣的同仁聯系交流(yangdu@cuhk.edu.cn)。
?
參考文獻
[1]?Hauser, A. S., Attwood, M. M., Rask-Andersen, M., Schioth, H. B. & Gloriam, D. E. Trends in GPCR drug discovery: new agents, targets and indications. Nat. Rev. Drug Disco. 16, 829–842 (2017).
[2]?Ye, N. et al. Orphan receptor GPR88 as an emerging neurotherapeutic target. Acs Chem. Neurosci. 10, 190–200 (2019).
[3]?Watkins, L. R. & Orlandi, C. Orphan G protein coupled receptors in affective disorders. Genes?11, https://doi.org/10.3390/genes11060694 (2020).
[4]?Lin, X. et al. Structural basis of ligand recognition and self-activation of orphan GPR52. Nature?579, 152–157 (2020).
[5]?Mizushima, K. et al. A novel G-protein-coupled receptor gene expressed in striatum. Genomics?69, 314–321 (2000).
[6]?Massart, R., Guilloux, J. P., Mignon, V., Sokoloff, P. & Diaz, J. Striatal GPR88 expression is confined to the whole projection neuron population and is regulated by dopaminergic and glutamatergic afferents. Eur. J. Neurosci.?30, 397–414 (2009).
[7]?Laboute, T. et al. The orphan receptor GPR88 blunts the signaling of opioid receptors and multiple striatal GPCRs. eLife 9, https://doi.org/10.7554/eLife.50519 (2020).
[8]?Meirsman, A. C. et al. Mice lacking GPR88 show motor deficit, improved spatial learning, and low anxiety reversed by delta opioid antagonist. Biol. Psychiatry 79, 917–927 (2016).
[9]?Quintana, A. et al. Lack of GPR88 enhances medium spiny neuron activity and alters motor- and cue-dependent behaviors. Nat. Neurosci. 15, 1547–1555 (2012).
[10]?Bi, Y. et al. The discovery of potent agonists for GPR88, an orphan GPCR, for the potential treatment of CNS disorders. Bioorg. Med. Chem. Lett.?25, 1443–1447 (2015).
[11]?Huang, W. J. et al. Structural insights into mu-opioid receptor activation.?Nature?524, 315–31 (2015).
[12]?Standfuss, J. et al. The structural basis of agonist-induced activation in constitutively active rhodopsin. Nature?471, 656–660 (2011).
[13]?Maeda, S. et al. Endogenous agonist-bound S1PR3 structure reveals determinants of G protein-subtype bias. Sci. Adv.?7, https://doi.org/10.1126/sciadv.abf5325 (2021).
?
?
文章轉自港中深醫學院生命健康學院微信公眾平臺,鏈接為https://mp.weixin.qq.com/s/XOFlMegnoUpxlk_C5d10BQ




