醫(yī)學(xué)院杜洋教授團(tuán)隊在Science等發(fā)表多篇創(chuàng)新藥物發(fā)現(xiàn)成果
醫(yī)學(xué)院杜洋教授團(tuán)隊近期在Science、Nature Commun.等合作發(fā)表多項高水平的創(chuàng)新藥物發(fā)現(xiàn)成果。過去三年來杜教授團(tuán)隊依托科比爾卡創(chuàng)新藥物開發(fā)研究院/冷凍電鏡等平臺,已經(jīng)以通訊作者在Cell、Science、Cell Research、Nature Commun.、Science Advances等發(fā)表多項生物醫(yī)藥的前沿工作,也體現(xiàn)了香港中文大學(xué)(深圳)在粵港澳大灣區(qū)乃至國內(nèi)生物醫(yī)藥大健康領(lǐng)域發(fā)展的積極勢頭和競爭力。
?
一、靶向腎上腺素能受體的無成癮鎮(zhèn)痛藥物開發(fā)
疼痛及阿片類藥物濫用的大流行凸顯了對新的非阿片類藥物治療疼痛的需求。許多非阿片類受體參與疼痛處理(傷害感知),但其中只有少數(shù)是已經(jīng)在治療上得到證實的鎮(zhèn)痛靶標(biāo)。α2A-腎上腺素能受體(α2AAR)是一種A類G蛋白偶聯(lián)受體(G-protein coupled receptor, GPCR),其在中樞神經(jīng)系統(tǒng)中的激活具有緩解疼痛的效果。已知的作用于α2AAR的藥物,例如咪唑類藥物clonidine (可樂定)和dexmedetomidine (右美托咪啶) ,具有鎮(zhèn)痛作用。不幸的是,它們還具有很強(qiáng)的鎮(zhèn)靜作用;這種鎮(zhèn)靜作用對這些藥物的主要適應(yīng)癥很重要, 但卻限制了它們在醫(yī)院中的作為鎮(zhèn)痛藥的廣泛使用。由于α2AAR能夠結(jié)合下游多種信號蛋白 (如G 蛋白, 抑制蛋白arrestin 等), 從而產(chǎn)生不同的生理效應(yīng) (如鎮(zhèn)痛、鎮(zhèn)靜作用), 因此開發(fā)一種結(jié)構(gòu)不同于可樂定、右美托咪啶等傳統(tǒng)咪唑類藥物, 并能選擇性 (偏向性) 激活下游特定信號通路的新型激動劑, 可能會消除或降低其鎮(zhèn)靜作用,從而作為非阿片類鎮(zhèn)痛藥而得到應(yīng)用。

(點(diǎn)擊圖片,閱讀文章)
香港中文大學(xué)(深圳)醫(yī)學(xué)院/科比爾卡創(chuàng)新藥物開發(fā)研究院杜洋教授,聯(lián)合埃爾朗根-紐倫堡大學(xué)Peter Gmeiner教授和美國加州大學(xué)舊金山分校Brian Shoichet教授團(tuán)隊等,共同通訊在國際一流科學(xué)期刊Science雜志(IF=63.71)發(fā)表了最新的研究成果:“Structure-based discovery of nonopioid analgesics acting through the α2A-adrenergic receptor”。斯坦福大學(xué)醫(yī)學(xué)院博士后/原科比爾卡研究院研究助理徐俊、加州大學(xué)舊金山分校博士生Elissa Fink、聯(lián)合埃爾朗根-紐倫堡大學(xué)博士Harald Hubner等并列第一作者。
該研究通過分子對接從3.01億個虛擬分子中篩選出了17個α2AAR的配體,其親和力低至12nM,其中許多配體為部分激動劑并且偏向Gi和Go信號通路。研究者通過單分子冷凍電鏡技術(shù)解析了其中兩種配體-α2AAR復(fù)合物結(jié)構(gòu),證實了分子對接的預(yù)測,并以此為模板進(jìn)一步優(yōu)化了配體結(jié)構(gòu)。’9087(最初篩選出的激動劑,中值有效濃度(EC50)52nM)和它的兩個類似物,'7075和PS75(EC50分別為4.1和4.8nM),在數(shù)個體內(nèi)疼痛模型中表現(xiàn)出鎮(zhèn)痛效應(yīng),同時沒有鎮(zhèn)靜的作用和脫靶效應(yīng)。這些新發(fā)現(xiàn)的激動劑展現(xiàn)出豐富的成藥潛力,并展現(xiàn)出沒有藥物依賴(相較于阿片類藥物)和鎮(zhèn)靜作用(相較于咪唑類藥物右美托咪啶等)的優(yōu)勢。
該工作主要揭示了三個重要發(fā)現(xiàn)。
首先,有效的激動劑能從大數(shù)據(jù)庫中直接篩選出,并可以和已知的激動劑無關(guān)。分子對接能鑒別具有體外活性的分子,通過結(jié)構(gòu)活性優(yōu)化使其有體內(nèi)活性。雖然直接能發(fā)現(xiàn)有效激動劑的情況很少見,但該實驗中對接實驗直接命中有效配體確實表明大虛擬篩選庫的優(yōu)勢,尤其是與已知配體不同的化學(xué)型的藥效。
其次,雖然’9087和其類似物’7075、PS75偏向Gi/o/z途徑并沒有β阻遏蛋白路徑活性,但是設(shè)計時并未以功能選擇性為設(shè)計標(biāo)準(zhǔn),這可能是因為它們新的化學(xué)型。雖然在其他研究中經(jīng)常出現(xiàn)類似情況,未表征的化學(xué)型會引起新的信號傳導(dǎo)途徑這一邏輯仍需更多研究。
第三,’9087和其類似物’7075、PS75不會在鎮(zhèn)痛劑量下引起鎮(zhèn)靜或運(yùn)動障礙,這有著廣泛的應(yīng)用前景,并且證明α2AAR激動劑能根據(jù)兩種效果被區(qū)分。

該研究從超大數(shù)據(jù)庫中對接篩選出了低EC50 α2AAR的部分激動劑,且與已知的配體無關(guān),讓之前未發(fā)現(xiàn)的配體受體相互作用賦予了新藥理。一些先前未經(jīng)表征的激動劑在神經(jīng)性和炎癥性疼痛模型中起到扛異常疼痛和鎮(zhèn)痛作用,并且能在正常動物中緩解急性傷害感受。最有前途的是’9087和PS75,它們都有強(qiáng)烈的鎮(zhèn)痛作用,沒有右美托咪定的鎮(zhèn)靜副作用,并且是可口服。雖然其對于心血管的副作用未作充分研究,這些藥物前體為新型非阿片類疼痛療法帶來新的曙光。
?
二、揭示大腦孤兒受體GPR88激活和別構(gòu)調(diào)節(jié)機(jī)制
G蛋白偶聯(lián)受體(G-protein coupled receptor, GPCR)是人類基因組中最大的膜信號蛋白家族,包含800多個成員。然而其中大約140個受體的內(nèi)源性配體仍然未被確定。這些孤兒受體(orphan GPCR, oGPCR)的配體結(jié)合和信號傳導(dǎo)機(jī)理尚不明確,例如自激活機(jī)制。因此,為oGPCR“脫孤”以及解析它們的結(jié)構(gòu)能夠幫助研究者進(jìn)一步了解GPCR的分子機(jī)理,為相關(guān)疾病的藥物開發(fā)帶來新的思路。GPR88隸屬于A類GPCR,在大腦(特別是紋狀體)中特異性表達(dá)。該受體能夠調(diào)節(jié)GABA和谷氨酸信號通路,以及包括多巴胺受體和阿片類受體等一些其他的GPCR活性。轉(zhuǎn)錄分析和小鼠敲除研究表明,GPR88在調(diào)節(jié)大腦和行為功能(例如認(rèn)知,情緒,基于獎勵的學(xué)習(xí)和運(yùn)動控制)中起著重要作用。因此,GPR88是治療中樞神經(jīng)系統(tǒng)相關(guān)疾病(包括精神分裂癥,帕金森氏病,躁郁癥,焦慮,抑郁癥和成癮)的潛在藥物靶標(biāo)。

(點(diǎn)擊圖片,閱讀全文)
近日,香港中文大學(xué)(深圳)醫(yī)學(xué)院、科比爾卡創(chuàng)新藥物開發(fā)研究院杜洋教授、柳正教授聯(lián)合埃爾朗根-紐倫堡大學(xué)Peter Gmeiner教授和日本東北大學(xué)Asuka Inoue教授團(tuán)隊,在國際一流科學(xué)期刊Nature Communication雜志(IF=17.69)發(fā)表了最新的研究成果:“Activation and allosteric regulation of the orphan GPR88-Gi1 signaling complex”。香港中文大學(xué)(深圳)為第一作者單位,杜洋教授為該研究最后通訊作者。陳耕博士、徐俊博士、Maximilian Schmidt和Asuka Inoue并列第一作者。
該研究基于冷凍電鏡技術(shù)(cryo-EM)解析了人類GPR88-Gi1和合成激動劑(1R,2R)-2-PCCA信號復(fù)合物的結(jié)構(gòu),表明了(1R,2R)-2-PCCA結(jié)合在由跨膜螺旋5、6的細(xì)胞質(zhì)末端和Gi1的α5螺旋C末端組成的變構(gòu)位點(diǎn),展示了正構(gòu)結(jié)合位點(diǎn)的電子密度代表未知的內(nèi)源性配體。MD和突變研究揭示了一組獨(dú)特的結(jié)構(gòu)特征和水介導(dǎo)的極性網(wǎng)絡(luò)的GPR88的獨(dú)特激活機(jī)制,為理解GPR88的配體結(jié)合、激活和信號傳導(dǎo)機(jī)制提供了一個結(jié)構(gòu)框架,并將促進(jìn)神經(jīng)精神疾病的創(chuàng)新藥物發(fā)現(xiàn)以及該受體的“脫孤”。
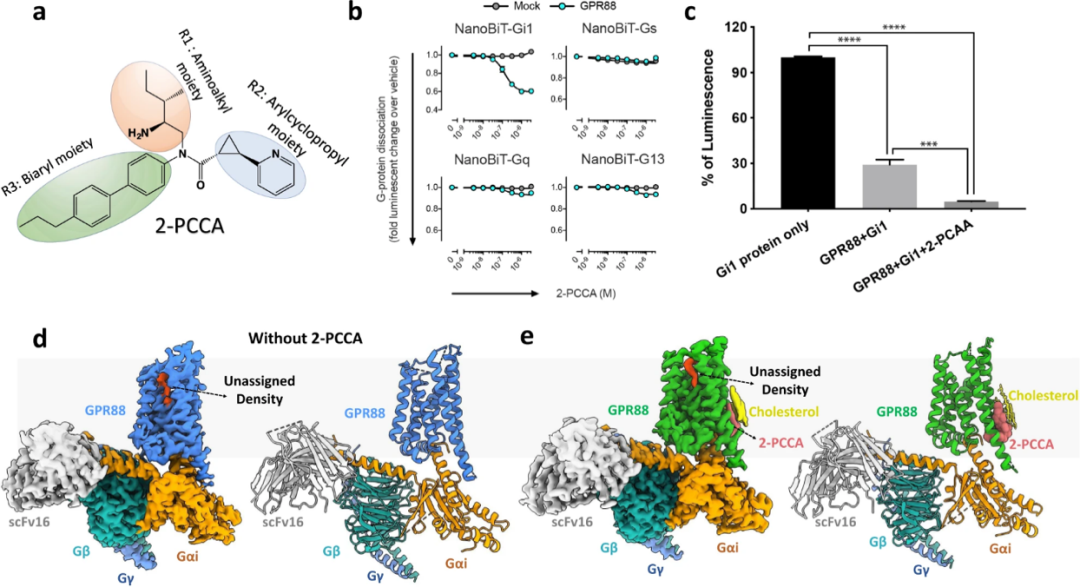
GPR88-Gi1冷凍電鏡結(jié)構(gòu)
該研究展示了結(jié)合或不結(jié)合合成激動劑(2-PCCA)的GPR88-Gi1信號傳導(dǎo)復(fù)合物結(jié)構(gòu)。這些結(jié)構(gòu)在GPR88的規(guī)范正構(gòu)袋中揭示了相似的電子密度,該密度可能代表受體的假定內(nèi)源配體。2-PCCA是一種變構(gòu)激動劑,與變構(gòu)位點(diǎn)結(jié)合,直接涉及與G蛋白的相互作用,進(jìn)一步穩(wěn)定了信號傳導(dǎo)復(fù)合物,從而促進(jìn)了GPCR88的高活性。Apo-GPR88-Gi和2-PCCA-GPR88-Gi的高分辨率結(jié)構(gòu)對比表明,正構(gòu)袋中的假定內(nèi)源配體可以與GPR88共純化。此外,未確定的密度和正位口袋的特性表明GPR88可能是響應(yīng)某些生物活性脂質(zhì)的受體。令人感興趣的是,與其內(nèi)源性激動劑S1P結(jié)合的鞘氨醇1-磷酸(S1P)受體的最新結(jié)構(gòu)顯示出與GPR88相似的正位結(jié)合口袋,口袋中形成了用于結(jié)合S1P的穿透性長隧道。因此研究者推測,GPR88的內(nèi)源性激動劑可能是一種與S1P相似的脂質(zhì)分子,并且該脂質(zhì)配體可能能夠與GPR88的變構(gòu)位點(diǎn)結(jié)合以調(diào)節(jié)信號傳導(dǎo)。但是,我們不能排除脂質(zhì)分子具有分支結(jié)構(gòu)和未確定的電子密度由靈活區(qū)形成的可能性。
總而言之,該研究提供了理解孤兒受體GPR88的配體結(jié)合,激活和信號轉(zhuǎn)導(dǎo)的結(jié)構(gòu)基礎(chǔ)。這些發(fā)現(xiàn)將促進(jìn)GPR88的“脫孤”。基于結(jié)構(gòu)的激動劑、拮抗劑設(shè)計可能會為中樞神經(jīng)系統(tǒng)疾病提供有價值的候選藥物。
?
三、揭示人體甲酰肽受體激活機(jī)制、助力新型抗菌抗感染藥物發(fā)現(xiàn)
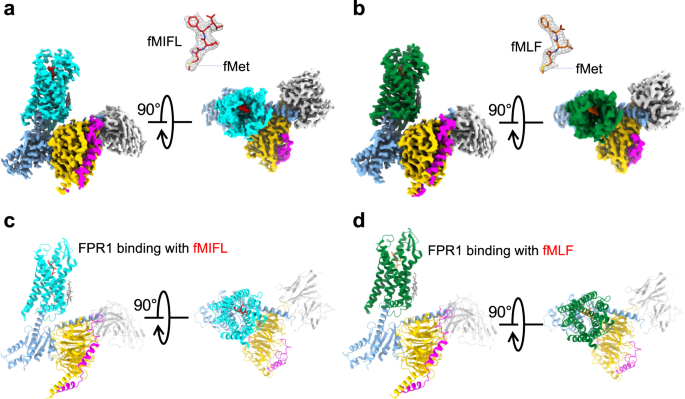
香港中文大學(xué)(深圳)醫(yī)學(xué)院、科比爾卡創(chuàng)新藥物開發(fā)研究院葉德全教授,聯(lián)合杜洋教授和胡紅麗教授在國際一流科技期刊Nature Communication雜志(IF=17.69)發(fā)表了最新的研究成果:“Structural basis for recognition of N-formyl peptides as pathogen-associated molecular patterns”。葉德全教授,杜洋教授和胡紅麗教授為共同通訊作者。杜洋教授團(tuán)隊陳耕博士為論文第一作者,王先坤、廖綺文、葛運(yùn)軍為論文共同第一作者。該研究通過冷凍電鏡技術(shù),分別解析了甲酰肽受體FPR1結(jié)合大腸桿菌甲酰三肽fMLF和金黃色葡萄球菌甲酰四肽fMIFL的結(jié)構(gòu),揭示了FPR1對N端甲酰基的具體識別機(jī)制,為今后以FPR1為靶點(diǎn)研發(fā)療效好副作用小的新型抗菌抗感染藥物提供結(jié)構(gòu)基礎(chǔ)。

(點(diǎn)擊圖片,閱讀全文)
甲酰肽受體FPR屬于Gi蛋白偶聯(lián)的A類GPCR,是一種重要的模式識別受體,與固有免疫系統(tǒng)中機(jī)體的防御密切相關(guān)。FPRs由三個成員組成,包括FPR1、FPR2和FPR3,主要通過偶聯(lián)G蛋白家族中的Gi/o異源三聚體介導(dǎo)信號傳遞。這些受體主要分布于中性粒細(xì)胞、嗜酸性粒細(xì)胞、巨噬細(xì)胞和樹突狀細(xì)胞等免疫細(xì)胞中,被甲酰肽激活后,一方面誘導(dǎo)中性粒細(xì)胞、肥大細(xì)胞等激活,釋放炎癥細(xì)胞因子,促使炎癥發(fā)生,同時,作為趨化物招募巨噬細(xì)胞等快速富集,引起活性氧的產(chǎn)生消滅病原微生物,并對病原體以及受損組織進(jìn)行吞噬清除,在免疫防御和調(diào)節(jié)過程起重要作用。原核生物如細(xì)菌的蛋白質(zhì)合成起始于被甲酰化的甲硫氨酸,這一特征是哺乳動物免疫細(xì)胞識別病原細(xì)菌的一個重要標(biāo)志,免疫細(xì)胞利用專門識別甲酰肽的受體FPR,發(fā)現(xiàn)追蹤入侵機(jī)體的細(xì)菌并將其殺滅。
本工作依托大學(xué)的冷凍電鏡平臺,解析了FPR1與高親和力配體fMLF和fMIFL及 Gi 蛋白的復(fù)合體結(jié)構(gòu),并根據(jù)結(jié)構(gòu)確認(rèn)了對甲酰肽識別和受體激活至關(guān)重要的獨(dú)特 R201XXR205 (RXXR) 序列。R201和 R205與 D106在甲酰基和 fMet 側(cè)鏈的識別中發(fā)揮關(guān)鍵作用,并能夠穩(wěn)定FPR1 的配體結(jié)合口袋。同時,該工作還在受體內(nèi)部發(fā)現(xiàn)了促進(jìn)甲酰肽結(jié)合的多個氫鍵作用力和疏水簇提供的疏水力,充分全面的闡述了FPR1識別和結(jié)合配體的分子機(jī)制。同時,這些結(jié)構(gòu)特點(diǎn)也得到了分子動力學(xué)模擬實驗的證明,進(jìn)一步說明了結(jié)構(gòu)的合理性和可信度。
綜上所述,我們利用單顆粒冷凍電鏡技術(shù)解析甲酰肽受體FPR1與Gi蛋白的高分辨率復(fù)合物結(jié)構(gòu),從而在原子層面上詳細(xì)闡釋了FPR1的配體識別及與G蛋白偶聯(lián)的機(jī)制。該項研究將促進(jìn)基于FPR1結(jié)構(gòu)的藥物研究,為新型抗菌抗感染藥物的研發(fā)提供結(jié)構(gòu)基礎(chǔ)。
?
四、揭示人體孤兒受體GPR17調(diào)控機(jī)制,助力神經(jīng)精神疾病藥物發(fā)現(xiàn)(封面文章)
多發(fā)性硬化癥、腦外傷、中風(fēng)和阿爾茨海默癥與中樞神經(jīng)系統(tǒng)的髓鞘損傷密切相關(guān)。研究表明,GPR17可以調(diào)控少突膠質(zhì)前體細(xì)胞分化成有髓鞘的成熟少突膠質(zhì)細(xì)胞,恢復(fù)髓鞘的功能。因此,GPR17可能是治療多發(fā)性硬化癥等神經(jīng)退行性疾病的潛在靶點(diǎn)。GPR17屬于A類G蛋白偶聯(lián)受體(G-protein coupled receptor, GPCR),主要在少突膠質(zhì)細(xì)胞中表達(dá)。GPR17激活后,通過與下游的Gi蛋白結(jié)合,抑制cAMP的產(chǎn)生,調(diào)控神經(jīng)元的代謝信號及生理功能。前期研究表明,GPR17有較高的“自激活”活性,但對這種“自激活”的原因和結(jié)構(gòu)基礎(chǔ)尚不清楚,限制了靶向GPR17小分子藥物的開發(fā)。

2022年9月10日,香港中文大學(xué)(深圳)醫(yī)學(xué)院杜洋教授團(tuán)隊在本土快速發(fā)展的主流期刊MedComm在線發(fā)表了最新的研究成果“Cryo-EM structure of G-protein-coupled receptor GPR17 in complex with inhibitory G protein”,并被選為封面文章重點(diǎn)報道。香港中文大學(xué)(深圳)杜洋教授為本文通訊作者,葉芳博士后,黃天仕研究員和陳耕副研究員為論文共同第一作者。香港中文大學(xué)(深圳)作為第一單位。
本研究首次解析了GPR17-Gi復(fù)合物的冷凍電鏡結(jié)構(gòu),揭示了GPR17的ECL2占據(jù)配體結(jié)合口袋,調(diào)控受體“自激活”的機(jī)制,為靶向GPR1的神經(jīng)系統(tǒng)退行性疾病藥物開發(fā)提供了重要的結(jié)構(gòu)基礎(chǔ)。

(點(diǎn)擊圖片,閱讀文章)
為解析GPR17的結(jié)構(gòu),杜洋課題組在昆蟲細(xì)胞SF9細(xì)胞共表達(dá)GPR17和Gi蛋白三聚體的復(fù)合物樣品,在未加入小分子配體情況下,通過冷凍電鏡技術(shù),解析了分辨率為3.02 ? GPR17-Gi復(fù)合物結(jié)構(gòu)。通過結(jié)構(gòu)分析,GPR17的ECL2巧妙的插入受體的正位結(jié)合口袋,與TM7(跨膜螺旋7),TM3(跨膜螺旋3)的氨基酸相互作用,激活GPR17。研究人員進(jìn)一步通過cAMP實驗驗證了ECL2上的關(guān)鍵氨基酸對受體的激活作用。通過對ECL2占據(jù)GPR17正位結(jié)合口袋分析,GPR17的內(nèi)源性配體可能是親水性分子,GPR17未來的“去孤兒化”研究可以考慮集中篩選中樞神經(jīng)系統(tǒng)中的親水分子。
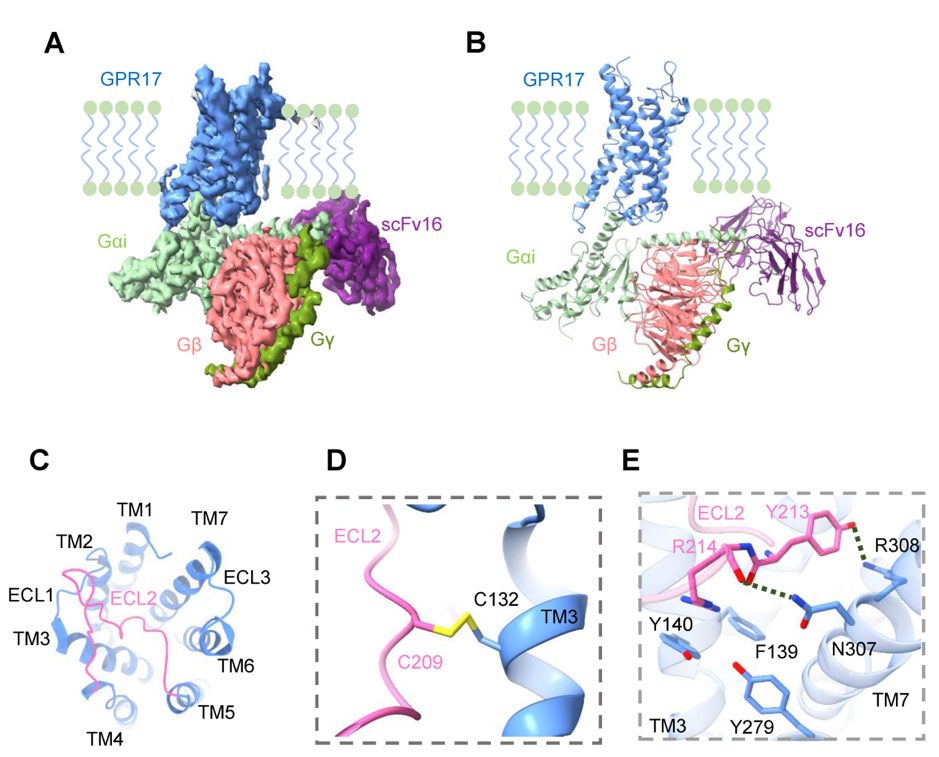
圖二 GPR17冷凍電鏡結(jié)構(gòu)及ECL2上的關(guān)鍵氨基酸與跨膜螺旋相互作用機(jī)制
本研究揭示了GPR17與下游信號分子Gi蛋白的相互作用機(jī)制。GPR17的TM3,Helix8和ICL3上的關(guān)鍵氨基酸參與了Gi相互作用界面,穩(wěn)定受體的激活構(gòu)象。這也為解釋GPR17與G蛋白結(jié)合,調(diào)控下游cAMP活性等信號通路提供了結(jié)構(gòu)基礎(chǔ)。
?
教授簡介

杜洋,國家級高層次人才,校長學(xué)者,深圳市優(yōu)秀教師。香港中文大學(xué)(深圳)醫(yī)學(xué)院/科比爾卡創(chuàng)新藥物開發(fā)研究院研究員。獲得中國科學(xué)技術(shù)大學(xué)博士學(xué)位后,赴美國斯坦福大學(xué)醫(yī)學(xué)院師從2012年諾貝爾化學(xué)獎得主Brian Kobilka教授從事博士后和研究科學(xué)家研究工作多年,獲得美國心臟協(xié)會全額獎學(xué)金和密歇根大學(xué)安娜堡醫(yī)學(xué)院助理教授職位。研究領(lǐng)域為G蛋白偶聯(lián)受體、冷凍電鏡和基于結(jié)構(gòu)的藥物設(shè)計等。杜教授已入選國家省市區(qū)等各級高層次人才計劃,并承擔(dān)各級多項科研項目開展前沿研究。
?
文章轉(zhuǎn)自香港中文大學(xué)(深圳)微信公眾平臺,鏈接為https://mp.weixin.qq.com/s/cU8VDEmGLwwG_fZXcBWaPA




